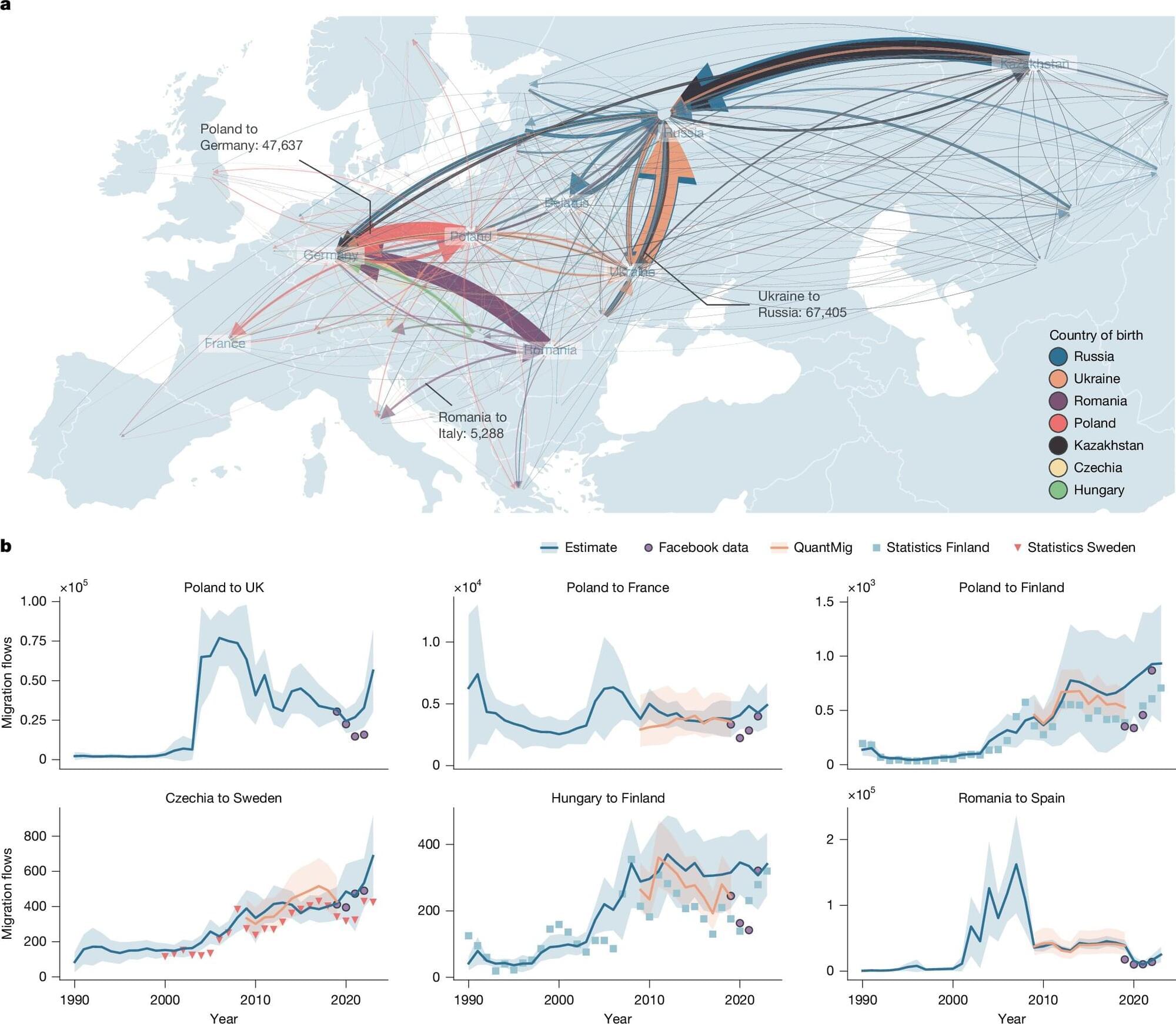
Global migration has risen sharply from approximately 13 million people per year in 2000 to around 35 million people per year in 2023. This is according to a new dataset on human migration published in Nature by researchers from the London School of Economics and Political Science (LSE), IIASA and the University of Hong Kong.
This rise in migration outpaces global population growth, showing a true per capita increase in human mobility. The trend is contrary to previous research efforts to quantify global migration flows.
Using deep learning, the researchers built the first dataset of migration flows between all countries for the period 1990–2023, offering a far more detailed picture of global movement than traditional data, which is highly fragmented.