From chemical building blocks, scientists have created synthetic cells that have most of the hallmarks of life.
Scientists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory and their collaborators have demonstrated a promising new approach for converting methane—the primary component of natural gas—into liquid chemicals that are precursors for many industrial chemicals and fuels. The research, described in a paper just published in Advanced Functional Materials, shows how molybdenum disulfide (MoS2), an earth-abundant industrial catalyst, can be used with minimal tweaking to selectively convert methane into methyl peroxide and other liquid oxygenate compounds at temperatures below 100°C (212°F). Methyl peroxide is a precursor for making methanol, an energy-dense liquid fuel that can be transported easily.
“The fact that this catalyst is an earth-abundant, domestically sourced material could change the game for converting natural gas into liquid chemicals,” said Brookhaven Lab chemist Sanjaya Senanayake, a corresponding author on the publication. “The catalyst achieves very high yields and high specificity for making important precursors for methanol and a wide range of other industrial processes.”
The project is part of a long-term strategy of the Catalysis: Reactivity and Structure group in Brookhaven Lab’s Chemistry Division to develop methane-conversion catalysts and processes. This group includes co-authors Senanayake, chemist Juan Jiménez and research associate Arephin Islam—all co-authors on the new publication.

Each unit of cost invested in Helicobacter pylori screening can generate approximately a fivefold return in gastric cancer prevention benefits.
The gastric cancer prevention research team at National Taiwan University Hospital and College of Public Health, National Taiwan University, has pioneered a globally applicable preventive model for gastric cancer control. To inform public health policymaking, the research team developed a globally adaptable decision-tree model to evaluate the cost-effectiveness of H. pylori screening. The findings were published in JAMA on June 1, 2026.
Building on Taiwan’s nationwide fecal immunochemical test-based colorectal cancer screening program, the gastric cancer prevention team has conducted a 10-year randomized clinical trial demonstrating that the additional use of an H. pylori stool antigen test (HPSA) alongside fecal occult blood testing could simultaneously achieve the dual goals of colorectal cancer and gastric cancer prevention. The findings were previously published on Sept. 30, 2024, in JAMA.
Plutonium is one of the most complex elements in the periodic table. First synthesized and isolated in 1940 by scientists at the University of California, Berkeley, plutonium has been studied closely for more than eight decades. It’s most often associated with its role in nuclear security, but it’s also vital to nuclear power, where it is produced in reactors and can be recycled as fuel. Despite plutonium’s importance, some of its most fundamental behaviors remain a mystery.
Scientists at the Idaho National Laboratory (INL) have made an important discovery: A compound called plutonium hexaboride (PuB₆) exhibits a one-of-a-kind quantum property known as a topological Kondo insulating state. Published in Physical Review Research, this finding marks one of only a handful of times such behavior has been observed in a plutonium material—opening a new window for research into how some of nature’s most complex elements actually work.
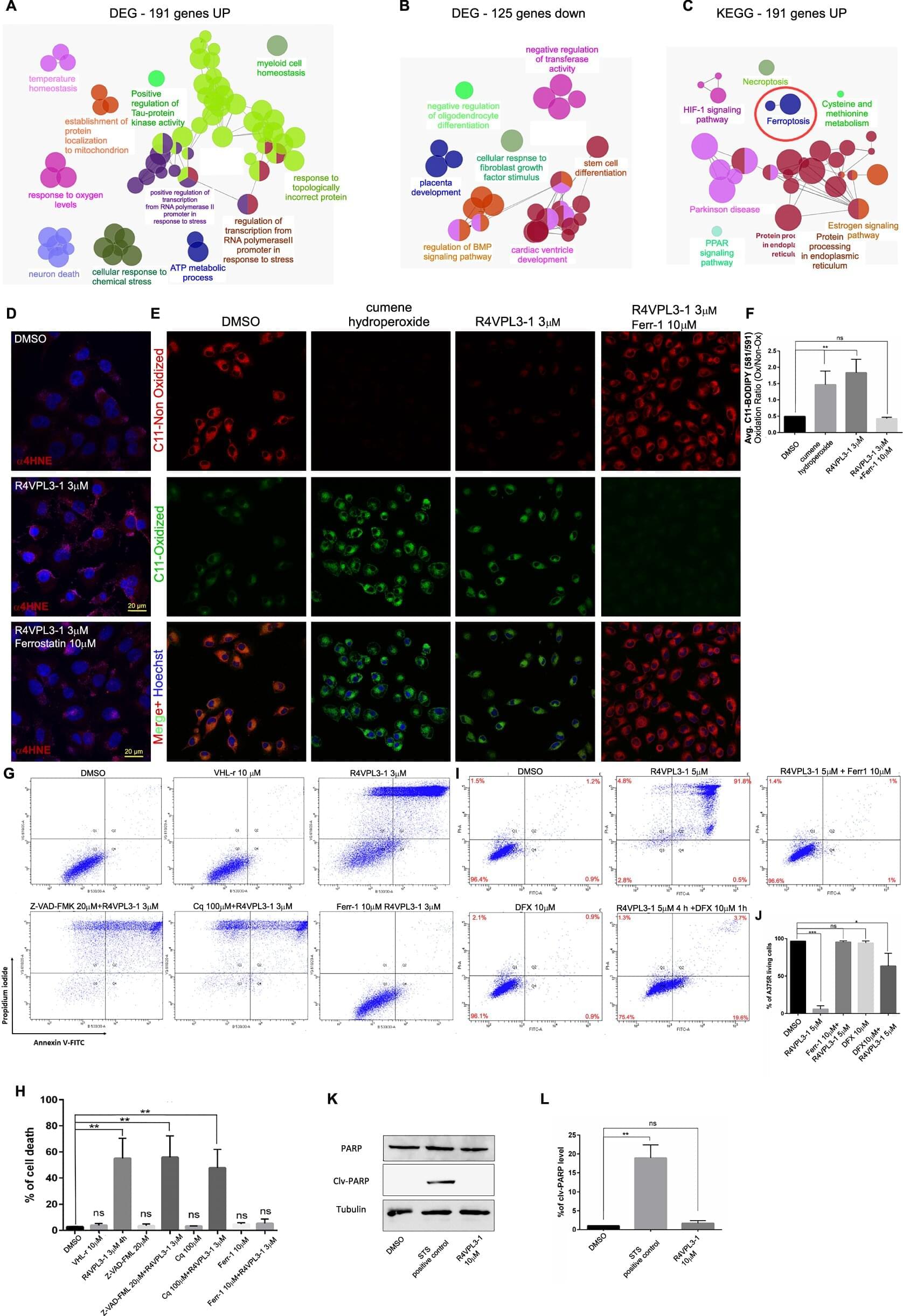
Researchers from two Technion faculties have jointly developed a new compound and demonstrated its effectiveness against aggressive tumor cells.
A study published in Oncogene presents an innovative strategy for the particularly complex medical challenge of destroying aggressive, treatment-resistant tumors.
The research was jointly led by early-career scientists Dr. Avital Oknin Vaisman and Dr. Deepanjan Panda from the laboratories of Prof. Amir Orian, head of the Rappaport Center for Cancer Research at the Technion-Israel Institute of Technology and a faculty member in the Ruth and Bruce Rappaport Faculty of Medicine, and Prof. Ashraf Brik of the Schulich Faculty of Chemistry.
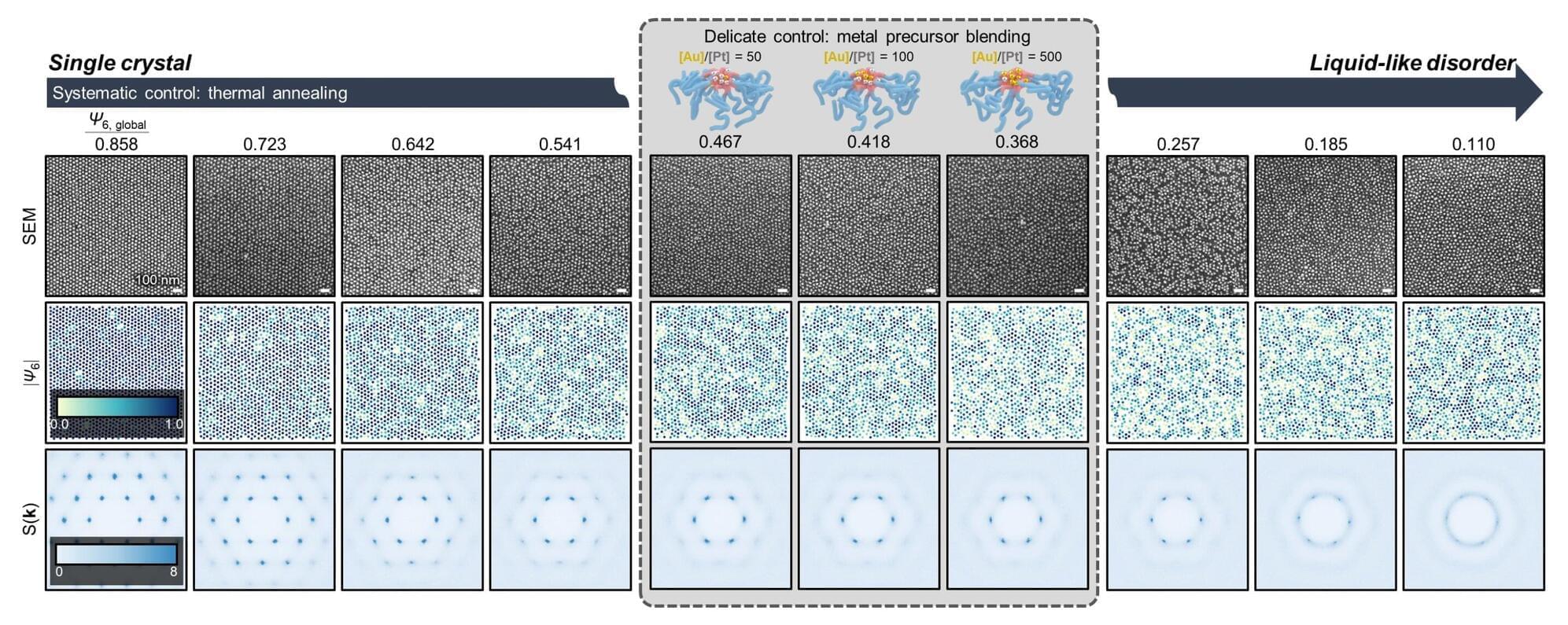
A research team has developed a methodology to precisely design and control the “degree of disorder” in nanopattern arrays using metal-infiltrated block copolymer (BCP) thin films. The work was led by Professor So Youn Kim of the Seoul National University College of Engineering Department of Chemical and Biological Engineering, in collaboration with Professor Su-Mi Hur’s team at DGIST and Professor S. Joon Kwon’s team at Sungkyunkwan University. The paper is published in the journal Nature Communications. The study was selected as an Editors’ Highlight in materials science and chemistry.
This disordered nanopattern fabrication technology is regarded as an innovative approach that enables precise control of nanoscale disorder structures—previously difficult to regulate—thereby opening new possibilities in the design of nano-optical and nanoelectronic devices.
In ordered structures, waves propagate over long distances, whereas in disordered structures, repeated scattering can lead to localization, where waves remain confined within a specific region. Such disordered structures exhibit unique functionalities that can induce localization phenomena for various types of waves, including light, sound and heat.

Saturn’s largest moon, Titan, is a unique environment in our solar system. It is the only moon (or body beyond Earth) to have a dense, nitrogen-rich atmosphere, and its methane cycle is very similar to Earth’s hydrological cycle, in which solid and liquid methane evaporate to form clouds and return to the surface as precipitation. In addition, its prebiotic surface environment and rich organic chemistry make it a prime destination for astrobiology missions, such as NASA’s Dragonfly mission (set to launch no earlier than July 2028).
And as Robert Zubrin said in his book, “Entering Space: Creating a Spacefaring Civilization,” Saturn’s moons could become the “Persian Gulf” of the solar system, with Titan a major one because of its rich resource environment. In a recent NASA-supported study posted to the arXiv preprint server, a team of researchers compiled an inventory of Titan’s resources and their potential use by future generations of humans. When comparing this satellite with other destinations (i.e., the moon and Mars), they conclude that Titan offers several potential benefits for human settlement.
The research was led by Conor A. Nixon, an astronomer and planetary scientist with the solar system Exploration Division (SSED) at the NASA Goddard Space Flight Center and the associate laboratory chief of its Planetary Systems Laboratory. He was joined by Ye Lu, a professor of aerospace engineering at Worcester Polytechnic Institute, and Jennifer E. Ruliffson, a professor of Materials Science and Engineering at the University of Florida. Their paper is under review for publication in Acta Astronautica.
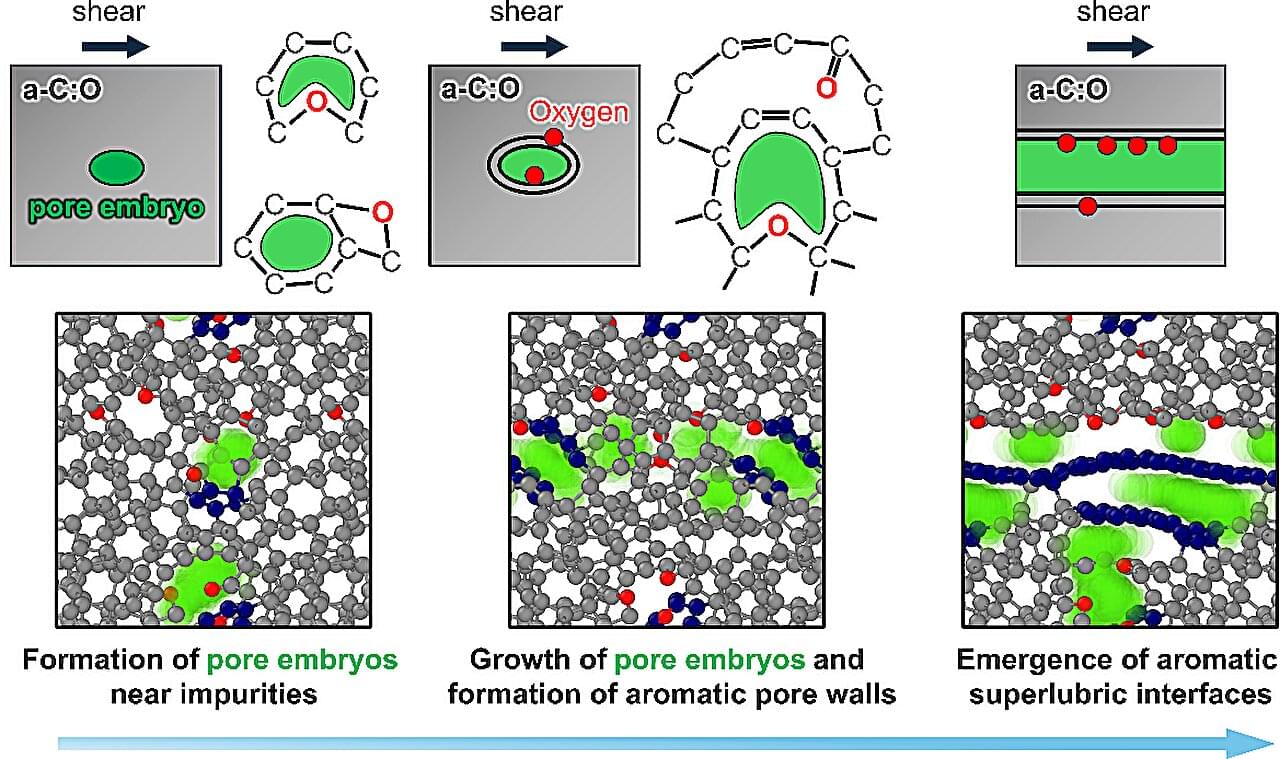
Engineers often treat impurities as a problem to eliminate to improve material performance. But new research from Osaka Metropolitan University and Fraunhofer Institute for Mechanics of Materials IWM suggests that in some cases, a little chemical messiness is exactly what helps materials slide more smoothly. The findings were published in Advanced Science.
When two surfaces slide or rub against each other, friction occurs. While friction is essential for many everyday applications, it also wears down machines, wastes energy and limits the lifespan of moving parts. Therefore, research has focused on achieving superlow friction, or superlubricity, in which surfaces can slide past one another with exceptionally low resistance.
“While graphene-or graphite-like structures are known to enable nearly frictionless sliding, creating and maintaining such structures in practical systems remains challenging,” said Takuya Kuwahara, lecturer at Osaka Metropolitan University’s Graduate School of Engineering and lead author of the study.

When the Lincoln Memorial Reflecting Pool turned green with algae just days after a US$15 million renovation, the U.S. government scrambled for chemicals and expensive technical solutions to fix the iconic landmark.
Trying to kill algae with chemicals is a common response when community ponds or other water features go green. But as a scientist who studies freshwater ecology, I can tell you there are better solutions that cost far less, last longer and carry less risk of harm to pets and wildlife.
Rather than battling against nature, these alternatives work with nature for long-term solutions.
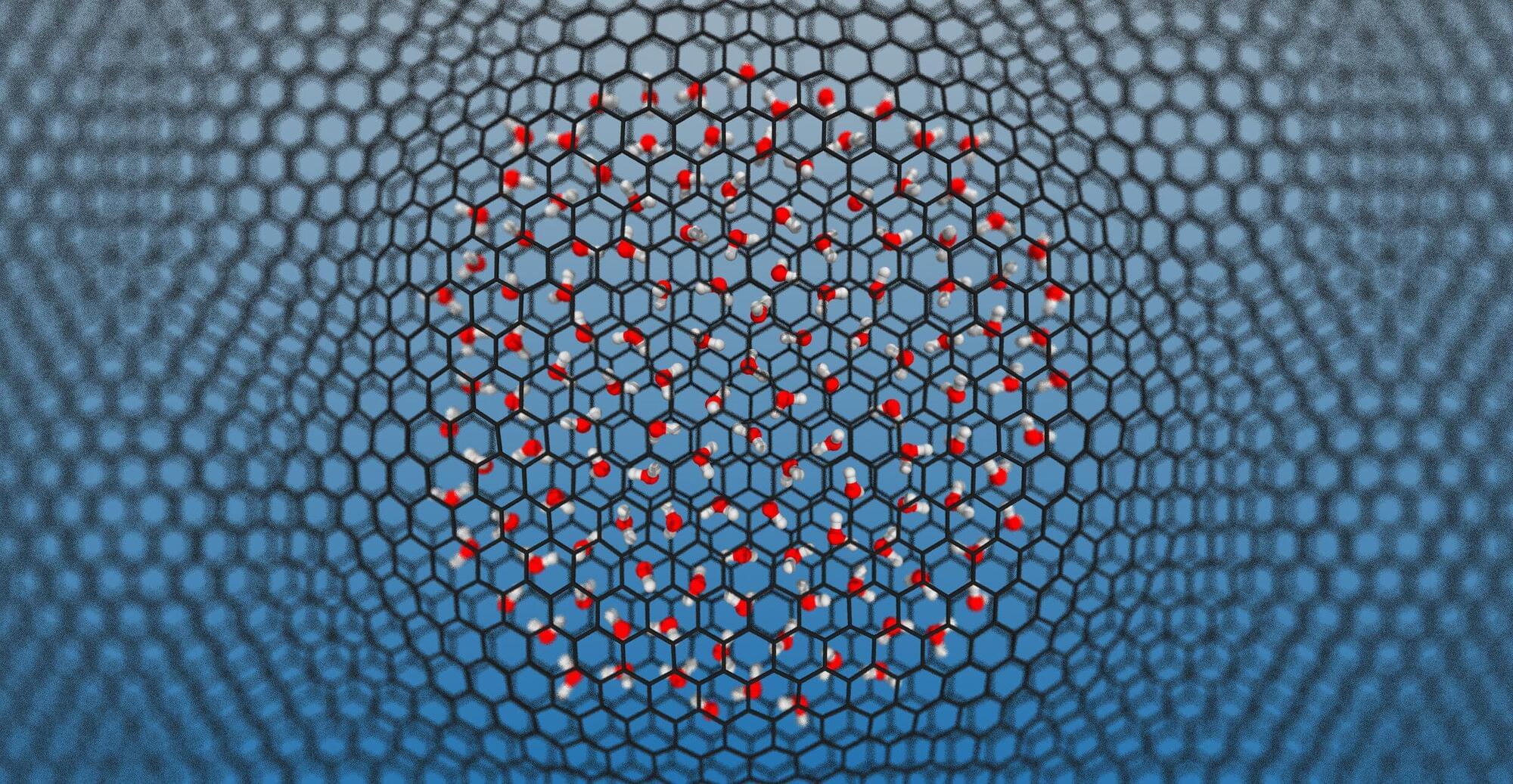
Water is the most studied molecule on Earth, yet a surprisingly basic question has gone unanswered for decades: When water is squeezed into gaps just a few molecules wide—as happens inside nanoscale pores, membranes and biological channels—does it become more or less chemically reactive?
This matters because water’s most fundamental chemical property is its ability to split into two charged species, H₃O⁺ (the hydronium ion) and OH⁻ (the hydroxide ion). This reaction defines the pH, a measure of how acidic or alkaline (basic) a solution is, and underpins all of acid-base chemistry, from how enzymes work in your cells to how electrodes function in batteries.
Through this research, the scientists wanted to understand whether (and how) confining water to nanometer-scale spaces affects this behavior.
{kind=link}