Today, more people are living past 100 than ever before — even though the maximum human lifespan hasn’t moved past 115 years. But is that about to change?
The Limit host Daniel T. Allen spent months talking to medical researchers, biohackers, and centenarians. He also went through a battery of tests worth over $12,000 at a longevity clinic to find out how long he might live.
In this episode, Business Insider looked into what could radically extend human lifespan, including FDA-approved drugs, cellular reprogramming, and Bryan Johnson’s $2 million \.
As an aging population experiences joint pain and inflammation at an all-time high, researchers at The University of Alabama in Huntsville (UAH), a part of The University of Alabama System, have published new findings suggesting continuous low-intensity ultrasound may help shift the body’s immune response from prolonged inflammation toward tissue repair, a discovery that could eventually contribute to novel treatments for joint injuries and post-traumatic osteoarthritis.
The study, published in Scientific Reports, was conducted by a multidisciplinary team of UAH researchers under the leadership of Dr. Anuradha Subramanian, a professor of chemical and materials engineering.
The work brought together biological experimentation conducted by Dr. Shahid Khan as part of his doctoral work with computational and statistical methods developed by Dr. Satyaki Roy, a professor of mathematical sciences, along with additional contributions from graduate student Owen Trippany.
After 300+ interviews on Singularity. FM, I ended up on the other side of the microphone.
Cadell Last invited me to Philosophy Portal and asked the questions that go all the way down. How a Bulgarian army nickname became “Socrates,” and why it started as an insult. How 300 resumes and one failed job interview accidentally started Singularity Weblog. And why, after 17 years of studying the technological singularity, I believe its biggest prophets got the most important thing wrong.
Ray Kurzweil is a genius and a genuinely humble human being. I’ve interviewed him and spent hours in his office. But his six epochs of the singularity converge into a single storyline where the universe literally wakes up. That is creationism in scientific clothing. It promises the same heaven of immortality and abundance, and it treats humanity as the chosen species.
Silicon Valley’s version is no better: the march of technology is inevitable, unstoppable, and there is nothing you can do about it.
That is not a prediction. That is a prison.
I grew up behind the Iron Curtain in Bulgaria. I watched the same technology build socialism in the East, democracy in the West, and fascism before both. The big choices are never technological. They are ethical, which is to say political.
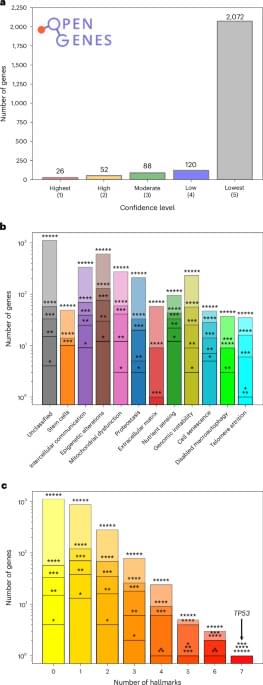
The authors introduce a network medicine framework showing that the hallmarks of aging form interconnected molecular modules in the human interactome. This new approach can help to identify existing drugs that might influence aging-associated transcriptional changes.
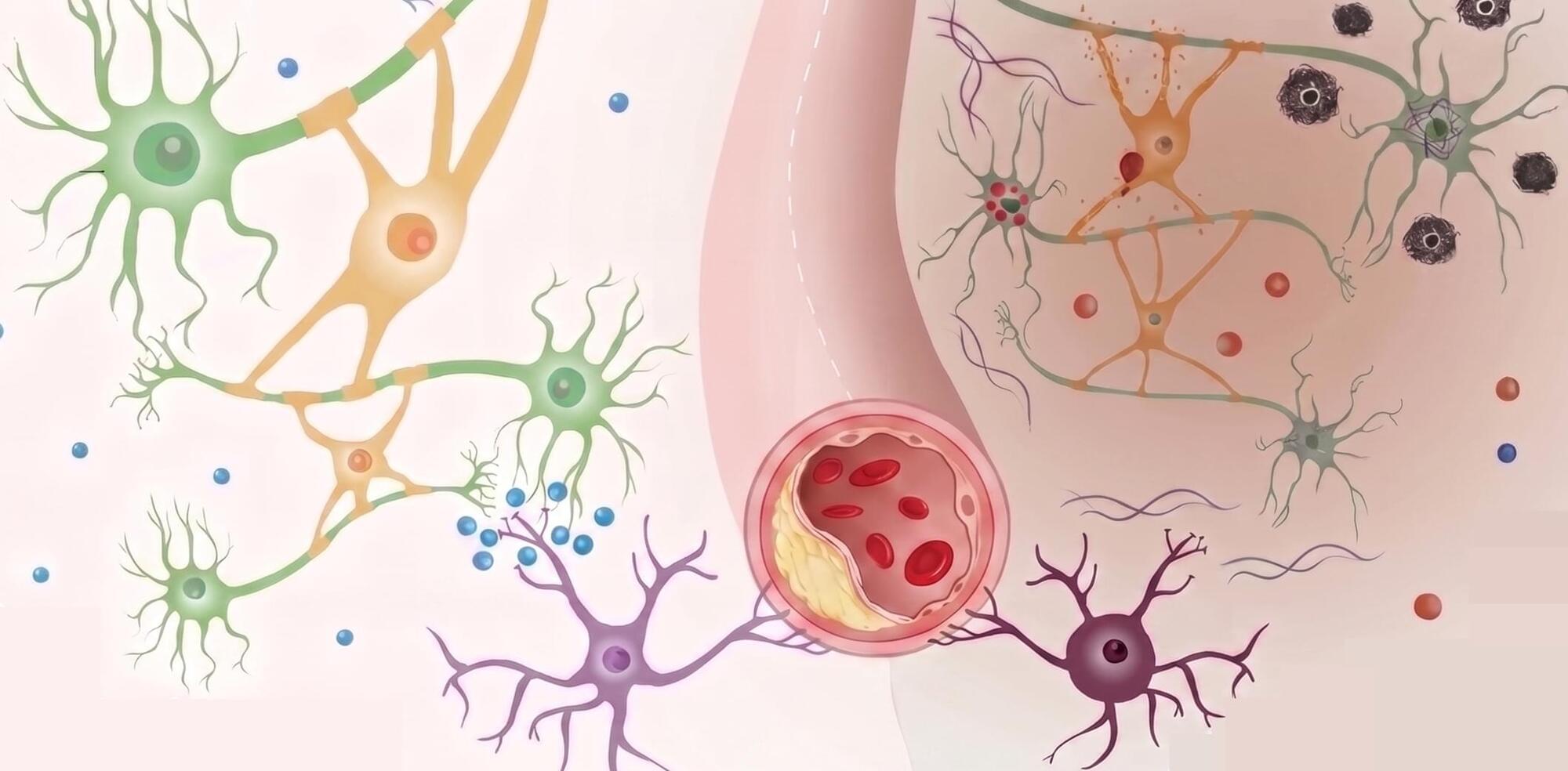
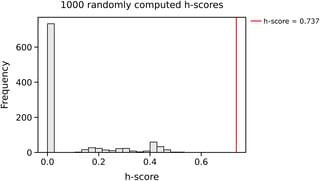
Senescence is a key manifestation of aging at the cellular level, caused by damage incurred by cells in time. In spite of their wide-ranging implications on how our multicellular bodies age, senescent cells are very challenging to identify due to their complex nature: many different aspects of cells are affected by this cellular state. This complicates defining clear criteria that help us decide whether a cell is senescent or not. In this paper, we propose a computational pipeline that enables us to identify a small subset of genes associated with senescence. The method combines two approaches commonly used in the study of networks, community detection and node centrality, and applies them to gene expression data obtained from the muscle tissue of mice after damage. The results obtained can contribute to establish the molecular correlates of a complex cellular state such as senescence.
Citation: Sabalic A, Moiseeva V, Cisneros A, Deryagin O, Perdiguero E, Muñoz-Cánoves P, et al. (2026) Cell-type resolved transcriptional network analysis of in vivo cellular senescence following injury. PLoS Comput Biol 22: e1014429. https://doi.org/10.1371/journal.pcbi.
Editor: Christoph Kaleta, Christian Albrechts Universitat zu Kiel, GERMANY.
Organ-specific age gaps showed strong associations with cancers affecting the corresponding organ. The strongest association was observed between kidney biological age and renal cancer (HR, 1.6). Organ-specific aging in lungs and intestines also increased the risk of lung cancer and stomach cancer, respectively (HR, 1.4 for both). The sensitivity analysis yielded largely similar results, except for attenuations in kidney and lung cancer, indicating the robustness of the primary findings.
The Global Proteomic Aging Clock predicted mortality from any cause as accurately as conventional risk factors. Combining the findings with established risk factors further improved mortality prediction compared with using risk factors alone.
Ultrasound guidance of filler injections is just one example of cosmetic dermatologists adjusting to deliver a more natural and satisfactory modification of aging skin.
Researchers from the University of Colorado Boulder, CU Anschutz, and Colorado State University have developed a set of experimental treatments that may help aging and damaged joints repair themselves in a matter of weeks. The therapies have shown promising results in animal studies, where they reversed signs of osteoarthritis and restored joint health.
The new approaches include a regenerative injection designed to be administered directly into a joint, as well as a biomaterial-based repair system that encourages the body’s own cells to rebuild damaged cartilage.
The work recently received a major boost from the federal Advanced Research Projects Agency for Health (ARPA-H), which announced that the team will move forward to the next stage of a project worth up to $33.5 million. The research is part of the ARPA-H Novel Innovations for Tissue Regeneration in Osteoarthritis (NITRO) program, led by ARPA-H Program Manager Dr. Ross Uhrich.





{kind=link}