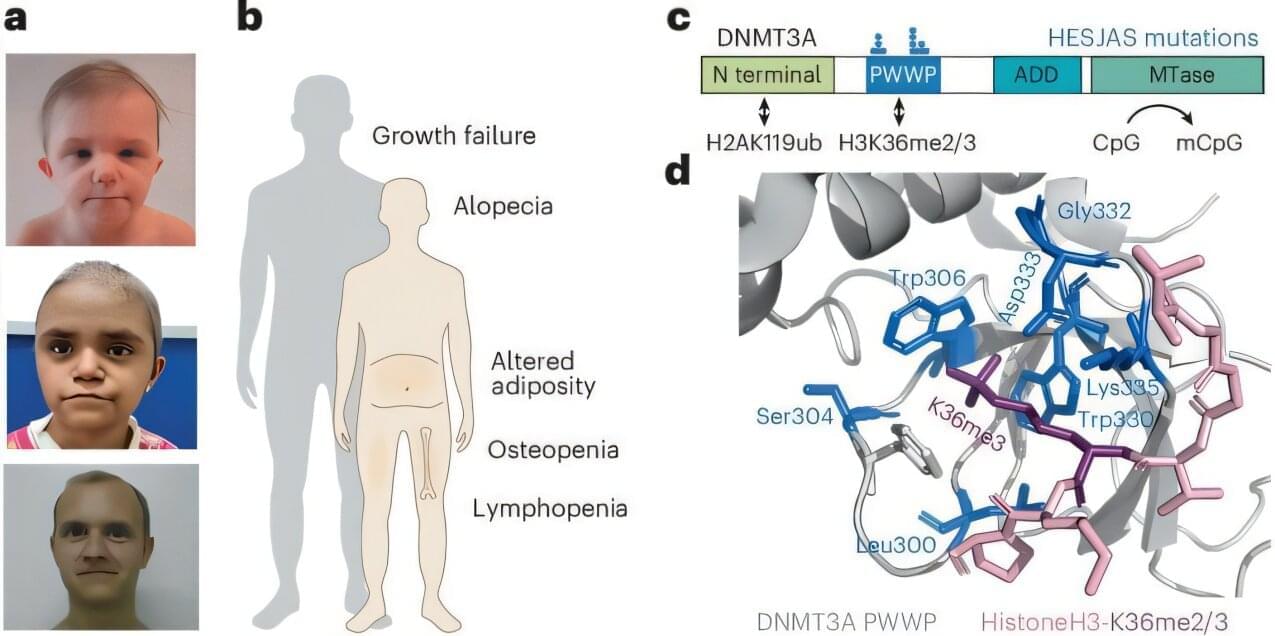
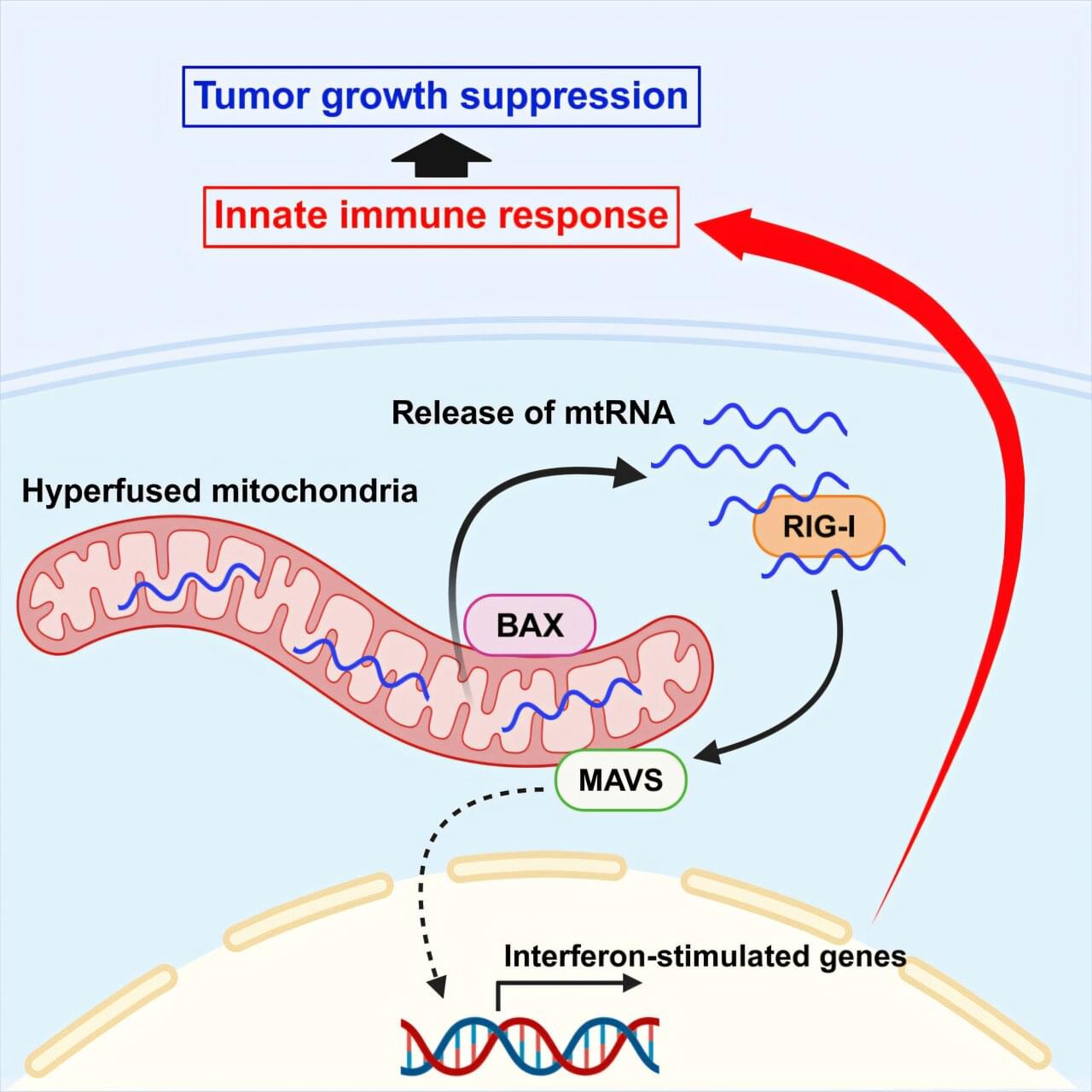
Human trisomy 21, responsible for Down syndrome, is the most prevalent genetic cause of cognitive impairment and remains a key focus for prenatal and preimplantation diagnosis. However, research directed toward eliminating supernumerary chromosomes from trisomic cells is limited. The present study demonstrates that allele-specific multiple chromosome cleavage by clustered regularly interspaced palindromic repeats Cas9 can achieve trisomy rescue by eliminating the target chromosome from human trisomy 21 induced pluripotent stem cells and fibroblasts. Unlike previously reported allele-nonspecific strategies, we have developed a comprehensive allele-specific (AS) Cas9 target sequence extraction method that efficiently removes the target chromosome. The temporary knockdown of DNA damage response genes increases the chromosome loss rate, while chromosomal rescue reversibly restores gene signatures and ameliorates cellular phenotypes. Additionally, this strategy proves effective in differentiated, nondividing cells. We anticipate that an AS approach will lay the groundwork for more sophisticated medical interventions targeting trisomy 21.
Keywords: CRISPR/Cas; Down syndrome; allele specificity; chromosome cut; chromosome loss; human trisomy 21.
© The Author(s) 2025. Published by Oxford University Press on behalf of National Academy of Sciences.