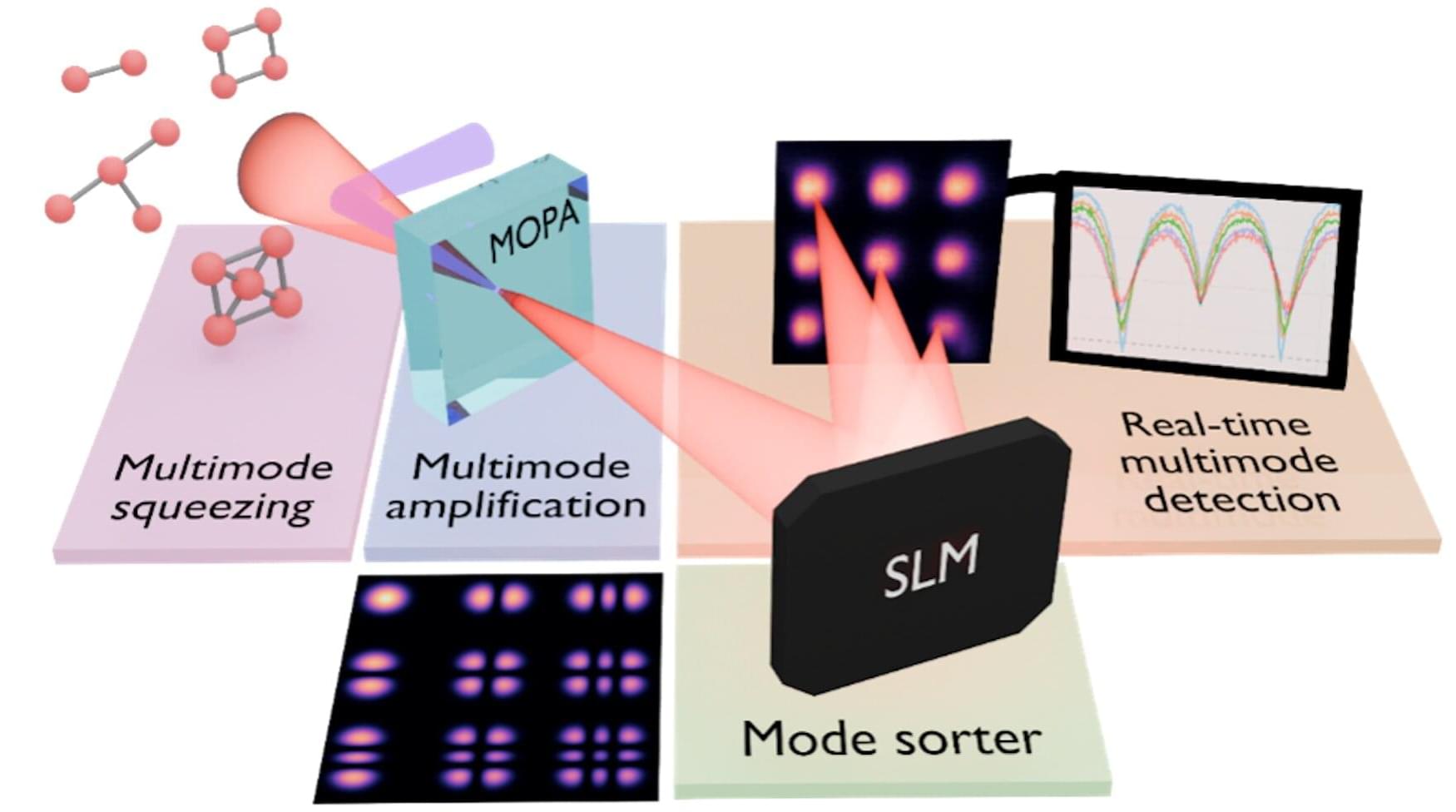
Quantum properties of light are extremely delicate. When researchers attempt to measure them, even small losses on the way to a detector can make them invisible, limiting their use outside carefully controlled environments. A collaborative team of researchers involving scientists at the Max Planck Institute for the Science of Light (MPL) has shown a new way to measure several quantum channels of light at the same time and reveal their entanglement, even when almost all of the light is lost before reaching the detector. The results, recently published in Nature Communications, open new possibilities for scalable quantum technologies.
Anyone who has used an old radio or television is familiar with noise in the sound or picture. These are random fluctuations that distort the transmitted information. Light behaves in a similar way. It also exhibits noise, appearing as fluctuations of the electromagnetic field. Even perfect laser light has such fluctuations, known as shot noise.




{kind=link}